In conjunction with Foresight Update 42
Recent Progress: Steps Toward Nanotechnology
By Jeffrey Soreff
|
|
|
|
|
|
|
|
Bernard Yurke, Andrew J. Tuberfield, Allen P. Mills Jr, Friedrich C. Simmel, and Jennifer L. Neumann, writing in [Nature 406:605-608 10Aug2000], describe the construction of a DNA actuator powered by complementary pairing between DNA strands.
| Top: The researchers assembled their tweezer motor from DNA with two double- stranded arms connected by a singled-stranded hinge, and two single-stranded “handles” at the ends of the arms. | 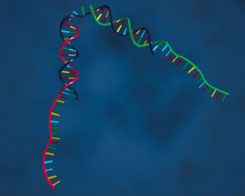 |
| Center: To close the tweezers, they add a special “fuel” strand of DNA | 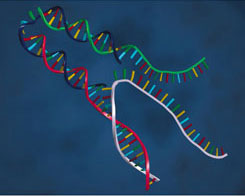 |
| Bottom: The fuel strand attaches to the handles and draws the two arms of the tweezers together. | 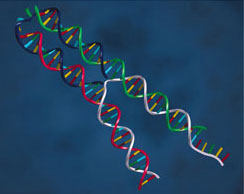 |
The authors’ machine consists of 3 permanent strands and one "fuel" strand. The 3 permanent strands consist of a central "A" strand and two peripheral "B" and "C" strands. Roughly half (18 bases) of the A strand is complementary to roughly half of the B strand, and the other half of the A strand (18 bases) is complementary to roughly half of the C strand. When the strands are assembled, both halves of the A strand are bound to halves of B and C, leaving roughly half (24 bases) of each of B and C floating free in solution. The A strand contains a hinge region formed by 4 unpaired bases between the regions that bind to B and to C. Single stranded DNA is much less stiff than double stranded DNA, so the portions of A that are bound to B and C act like a pair of rods extending from the hinge region, forming "tweezers" roughly 7 nm long.
The action of the "F" ("fuel") strand is to bind to the free halves of B and C, pulling the free ends of the rod-like A-B and A-C regions together. F is divided into two 24 base halves (plus an extra 8 base overhang), with one half binding to the free half of B and the other binding to the free half of C. The authors found that the ends of the tweezers were 6 nm closer together with the F strand present than with it absent.
The authors detected the movement of their tweezers by labelling the ends of the A strand with two fluorescent dyes, "TET (5′ tetrachloro-fluorescein phosphoramidite) and TARA (carboxy-tetramethylrhodamine)." When the two ends of strand A are far apart, (when the tweezers are open) TET fluoresces strongly when illuminated. When the two ends of A are close together (when the tweezers are closed), illuminated TET transfers its energy to TARA and fluorescence is suppressed.
One problem with this actuator is the formation of dimers. In normal operation F closes the tweezers by binding to B and C in the same actuator. Unfortunately, F can also bind to B and C strands from different actuators, tying them together into dimers or larger structures. The authors estimate that "about 80%" of their tweezers are closed rather than dimerized. The authors note that "dimer formation could in principle be avoided completely by tethering the tweezers to a solid substrate to prevent interaction."
The authors repeatedly cycled their actuators, opening and closing them 7 times. Opening the actuators is not just a matter of removing the F strands from solution and letting them diffuse away. Each base pair adds about 3 kT to the binding between strands, so the total binding energy is roughly 144 kT, sufficient to preclude desorption even on geological time scales. Instead, the authors add a complementary F’ strand to the solution. The F’ strand binds to the F strand more strongly than F binds to B and C, because F’ also matches the 8 base overhang region of F as well as to the 48 bases binding F to B and C. The F’ strand pulls the F strand off the actuator base by base (by "branch migration"), so few bases are unpaired at any point in the process. Both closing and opening the tweezers take approximately 13 seconds.
The authors write that "because the binding between fuel and machine is sequence-specific, the DNA strands that act as fuel may also serve as information carriers to coordinate components of a complex machine or to carry signals between machines." The specificity allows many (nominally up to 424 ~2.8 x 1014 for 24 base binding sites) similar actuators using the same general design to be independently controlled by their different fuel strands. Perhaps sequence-dependent DNA actuators can be combined with Nadrian Seeman’s DNA polyhedra to yield multiple degree-of-freedom manipulators, such as Stewart platforms, albeit initially ones with binary actuation of each degree of freedom.
For further information available on the Web (courtesy of Nanogirl News)
Carbon nanotubes, atomically smooth cylinders of graphite, have prompted a great deal of interest over the last decade due to their remarkable mechanical and electronic properties. The three clusters of papers summarized in this section describe advances in the application of these materials to memories, to electromechanical switching, and to the fabrication of low friction bearings.
Thomas Rueckes, Kyoungha Kim, Ernesto Joselevich, Greg Y. Tseng, Chin-Li Cheung, and Charles M. Lieber, writing in [Science 289:94-97 7July2000], describe calculations about and experiments with crossed nanotube storage elements. The authors have used the Van der Waals attraction between two closely spaced perpendicular nanotubes to give the pair two stable mechanical states. In one state the tubes are well separated and unstrained, while in the second they are in close contact and the attraction between them bends the upper tube, holding them together. These devices remind me of mercury-wetted reed relay contacts, with similar hysteresis from contact forces. The authors were able to both switch between the states and sense them electronically. While the switching between states was done electronically, the actual storage mechanism requires no applied voltages and is nonvolatile. The authors calculate potential switching rates of 100GHz and densities of 1012 bits/cm2
The authors’ memory array design has two perpendicular arrays of nanotubes, one array above the other, with a crossbar of memory devices formed by their intersections. If the design contained only ohmic switches between the layers, then the bits could not be sensed independently. For example, if bits are on, forming connections, at positions {3,10}, {12, 10}, and {12, 17} in the array, then there would appear to be a connection at {3, 17}, even if that bit is supposed to be off. The "standard solution to this problem is the incorporation of diodes at each cross point." This prevents the backwards flow of current that would otherwise cause the unwanted connections. The authors consider the ideal solution to be using contacts between metallic nanotubes in one layer with semiconducting nanotubes in the other to effectively form Schottky diodes at each memory element. They also suggest some other approaches.
The authors calculated the properties of their memory element using (10,10) nanotubes. Their calculated densities are set by the balance between the bending stiffness of the nanotubes and the strength of the attraction between them. The minimum size needed to make a weak enough spring for bistability is 10 nm on hard substrates and 5 nm on soft ones. The 100GHz switching speed includes time for both mechanical switching and for RC charging of the array.
Experimentally, the authors built junctions from 50 nm diameter nanotube ropes using a micromanipulator. One junction could be switched on by 10 volts between the tubes, and off by 40 volts applied to both tubes. The on resistance was 140 megohms, while the off resistance was 1.4 gigohms. This device could be switched repeatedly for several days. Another device had an on resistance of 112 kilohms. The assembly of an array of these devices has not yet been shown experimentally.
This memory device is a high performance application of nanotechnology. It is potentially comparable in density to some of the mechanically scanned AFM memory proposals, but has a much higher potential speed. It would also put a very high density of independent mechanical actuators under electronic control, which might be useful in driving nanoscale robotics.
Two groups of researchers have modified the electronic properties of nanotubes by bending them mechanically. A group mostly at Clemson: D. Tekleab, R. Czerw, D.L. Carroll, & P.M. Ajayan, writing in [Appl.Phys.Let. 76:3594-3596 12Jun2000], permanently bent multi-walled nanotubes (MWNTs) ultrasonically and saw "local metallic character" at the kink. A group mostly at Stanford: Thomas W. Tombler, Chongwu Zhou, Leo Alexseyev, Jing Kong, Hongjie Dai, Lel Liu, C.S. Jayanthi, Meijie Tang, & Shi-Yu Wu, writing in [Nature 405:769-772 15Jun2000], reversibly bent single-walled nanotubes (SWNTs) with an AFM tip and saw hundredfold drops in conductivity in their experiments.
The (primarily) Clemson group dispersed their MWNTs in tetrahydrofuran, then applied 200 watts of ultrasound to the dispersion. They found that tubes with diameters <5nm "are more easily kinked while a majority of the larger tubes remained undeformed." The authors performed STM imaging and spectroscopy in UHV with a Pt-Ir tip on a HOPG substrate.
Spectroscopy measurements from I(V) measurements from one tube with a 25 degree kink were presented. The tube was originally semiconducting, with a 0.2 eV band gap far from the kink on both sides. Near the kink "the band gap close to the Fermi energy has become narrower by the addition of interface states to the gap."
The authors interpret these states as being locally "metallic", "analogous to a quantum dot". They consider local strain, topological defects, and interlayer interactions to be possible causes for the interface states. They find that the changes to the electron energy spectra of the tube fade out beyond ~1.7 nm from the kink, confirming that these states are local.
The (primarily) Stanford group measured conductivity in a SWNT while bending it with an AFM tip. This group suspended SWNTs over trenches etched into a SiO2 surface. They describe experiments on a 3.1 nm diameter SWNT, suspended over a trench 605 nm wide and 175 nm deep. The authors grew the SWNTs using patterned catalyst islands. They contacted them electrically by depositing micron-wide electrodes of "20 nm thick Ti and 60 nm thick Au" on them, one on each side of the trench.
The authors measured conductance through their SWNTs while pressing on the portion of the tube that crossed the trench. The tube was repeatedly pressed with an AFM tip, and both force and conductance were measured as a function of the SWNT’s deflection down into the trench. For small deflections the force was proportional to deflection3, as was expected for an initially unstressed elastic string.
The resistance rose dramatically for large deflections, from 200 kohms for the undeflected SWNT to 25 megohms for a deflection producing a 14 degree bend and an average strain of 3%.
Both the forces and the resistance changes were almost perfectly reversible, and repeatable from cycle to cycle. The authors use this to rule out damage to the metal-SWNT contacts and motion of the SWNT along the substrate as contributors to these effects.
The authors did quantum mechanical calculations for a (5,5) SWNT subjected to pressure from a tip while supported from its ends. It also showed a drop in conductivity, which was interpreted as due to the formation of additional carbon-carbon bonds between the front and back of the tube. These bonds tied up some of the orbitals that form the conducting pi states in the unstressed SWNT. The local hybridization of the carbon shifted from conducting, unsaturated sp2 towards insulating sp3. These are different from previous calculated results, "in which the nanotube structure is more or less uniformly bent or strained" (because the bending was produced by tilting the ends, rather than applying force from a local tip). The previous results predicted smaller changes in hybridization, and therefore smaller changes in conductivity than those in this paper.
The sharper changes in hybridization than previously expected should assist in selective mechanochemistry, perhaps allowing a sequence of substituents to be chemisorbed onto a SWNT surface.
This work is also a strong indication of the potential value of multi-tip scanning probe experiments. Even the most straightforward dual tip setup, one where the tips approach from diametrically opposed directions, would be a great boon to this work. One could measure changes in geometries, in local strain, and in I(V) spectra on the side of the tube opposite the AFM tip. One might be able to directly demonstrate the local hybridization changes from spatially resolved I(V) spectra, perhaps with atom-by-atom resolution.
For further information available on the Web:
John Cumings and A. Zettl, writing in [Science 289:602-604 28Jul2000] describe experiments on the use of multi-walled nanotubes (MWNTs) as linear bearings.
| A computer-generated image of nested multi-walled carbon nanotubes (MWNTs), as described in the work by Cummings & Zettl. |  |
The authors’ basic observation was that they were able to "spot-weld" a carbon-tipped manipulator to inner layer(s) of a MWNT, then slide them back and forth with respect to outer layer(s) of the MWNT. They observed these motions inside a TEM, recording the motion on video. The MWNT shown in their TEM images "originally had 9 walls, with an outer diameter of 8 nm and an inner diameter of 1.3 nm." The inner set of tubes that they exposed and attached to was "4 nm in diameter (consisting of four concentric walls)."
They pulled the core tubes out to an extension of 330 nm. They repeated the motion "about 20 times for several different MWNTs", and found that, even "at the atomic level", no changes were produced in the structures of the tubes. They "conclude that the nanotube sections are near perfect sliding surfaces, apparently free from wear for all cycles."
Unfortunately, while this conclusion is consistent with their experimental evidence, they have not ruled out processes that cause defect formation with some probability of much less than 5% per cycle. It would be more convincing if they could, for instance, do a temperature dependence study at temperatures high enough that thermomechanical damage was observable, observe the (presumably Arrhenius, Ae-E/kT) temperature dependence, then extrapolate back down to ambient temperature.
The authors also observed the behavior of their MWNTs on breaking the weld to the manipulator (by fatiguing the weld with "lateral deflections of the manipulator"). After the weld was broken, the inner MWNT layers retracted into the outer layers within a single video frame (33 milliseconds). From surface energy calculations, they conclude that force retracting the core nanotubes back into the outer tubes was 9 nN, and that retraction took 4.6 nanoseconds.
The authors reason that frictional forces must be less than the Van der Waals forces pulling the core nanotubes back into their sheath. They conclude that "recent atomic-scale frictional force measurements using conventional materials yield [frictional force] values about three orders of magnitude greater than the upper-limit frictional forces found here for MWNT surfaces."
In the same issue of Science (pp. 560-561) Laszlo Forro comments on these experiments, noting that they confirm Eric Drexler’s calculations of very low frictional forces between atomically smooth, incommensurate surfaces. Forro also suggests that nanotube cores could be extruded with electrostatic forces, essentially producing an electrostatic solenoid. If so, in addition to being a useful component, this would allow experiments to more closely confirm the magnitude of the retraction force and of frictional forces in this structure.
In [Nature 406:586 10Aug2000], John Cumings, Philip G. Collins, and A. Zettl describe how they exposed the inner layers of a MWNT, an essential step in their bearing experiments. In short, they zap them electrically. A MWNT "is brought into intimate contact with the tip of the nanotube at 2.9 V and 200 µA." This peels away layers from near the tip, leaving a stepped profile. In their TEM images, the stepped area extends about 140 nm from the tip of their (originally 12.6 nm in radius) MWNT. They are able to repeat the procedure, trimming their MWNT down to a radius of 2.1 nm.
The authors do not attribute the removal of the outer layers of their MWNT to Joule heating, but rather to "highly localized dissipation at defect scattering sites, located primarily at the ends of the tube." They attribute the selective removal of the outer layers of the MWNT to localization of current flow in these layers.
This demonstration of low friction between MWNT layers is valuable confirmation that they will be useful mechanical components in molecular nanotechnology. Over the short term, isolating or synthesizing MWNTs with consistent indices, not in one, but multiple layers, allowing consistent mechanical properties and attachment techniques, should be a challenging project.
For further information available on the Web:
Proteins are important potential components in early molecular nanotechnology. They fold into a rich variety of 3D structures.
Giuseppe Zaccai has written a short review paper in [Science 288:1604-1607 2Jun2000] describing the use of neutron scattering to probe protein dynamics.
Neutron scattering has been used to quantify thermal positional disorder in proteins. The thermal fluctuations in positions of the atoms in a sample can alter the directions and energies of scattered neutrons. Their mean square scatter from their equilibrium positions "are calculated from the angular dependence of the scattered elastic incoherent intensity." A major advantage of incoherent scattering is that "samples need not be either crystalline or [even] monodisperse". Incoherent neutron scattering is also insensitive to static disorder in samples, which in coherent x-ray diffraction becomes combined with the thermal movement. Neutron scattering is also complementary to x-ray diffraction because the former is very sensitive to hydrogen atoms, which are nearly invisible to x-rays. On the other hand, since neutron scattering cross-sections are considerably smaller than those for x-rays, "~200 mg of material [is] required for a neutron-scattering experiment."
These studies, amongst other techniques, have found "dynamical transitions from harmonic to anharmonic regimes … at ~200K (-73o C) in various proteins." In myoglobin, for example, there is a transition at ~200K from a "harmonic" regime where all the atoms are trapped in single potential wells to an anharmonic regime where jumps between wells become important. The anharmonic regime is important for the biological function of myoglobin (O2 binding), but may cause problems for use of proteins as mechanical elements in nanotechnology. In the harmonic regime the protein acts like it has a consistent potential well. In experiments on hydrated myoglobin powder, for instance, the scatter is consistent with a 2 N/m spring constant. The mean square scatter in position increases linearly with temperature in this regime.
In the anharmonic regime the atoms start to jump between potential wells, so the scatter starts to increase faster than in the harmonic regime. The effective spring constant drops quite dramatically, to 0.3 N/m in myoglobin. There is considerable evidence that "conformational flexibility is essential for enzyme catalysis and for biological molecular activity in general." In the case of myoglobin, for instance, the static x-ray diffraction structure provides no path for an O2 molecule to diffuse into the molecule and reach the heme binding site. Only fluctuations in the structure permits its biological function of binding O2. Note that these fluctuations start to appear at a temperature where myoglobin is far from denaturing. Evidently typical native, well-folded proteins have 3 well separated states as temperature increases:
While inter-well jumping is necessary for biological function, it implies that even a well-folded protein is not a eutactic structure from the viewpoint of molecular nanotechnology. The covalent bonds aren’t breaking, but it seems likely that hydrogen bonds which help hold together the 3D structure do break transiently. In other words, the drop in effective stiffness that happens on entering the anharmonic regime makes proteins significantly worse at maintaining positional control in the presence of thermal noise. Ideally, we would prefer structures that are as stiff as possible, except along one or two carefully chosen mechanical degrees of freedom.
Fortunately, this paper also describes experiments with trehalose, a disaccharide that some organisms use to survive dehydration. It "appears to help proteins avoid denaturation by surrounding them with a continuous vitreous layer." When trehalose was added to myoglobin, the protein was held in the harmonic regime to at least 310K. The trehalose also raised the effective spring constant of the myoglobin to 3 N/m. Since trehalose prevents denaturation, presumably the secondary and tertiary structure of myoglobin is left intact. That suggests that the hydrophobic core of the protein is not directly held in place, but rather that edge effects from the trehalose are sufficient to stabilize it.
Overall, the impact on nanotechnology appears to be that designs incorporating protein components must either operate at moderately low temperatures, glaze their proteins with a high-melting hydrophilic shell, or tolerate transient scission of hydrogen bonds in their protein components.
D.X. Shi, L.P.Ma, S.S. Xie, and S.J. Pang, writing in [J. Vac. Sci. Technol B. 18:1187-1189 May/June 2000], describe writing 0.8 nm (presumably diameter) conductive marks in a thin organic film with an STM, improving on their previous 1.4 nm marks. The marks were stable for at least the 2 hour scanning session. They attribute the marks to polymerisation of the film under the STM tip.
In this paper, the authors’ organic film is 3-phenyl-1-ureidonitrile, C6H5-NH-CO-NH-CN. They vacuum evaporated a 10 nm thick layer of this compound on to a HOPG (highly oriented pyrolytic graphite) substrate. They wrote their conductive marks under ambient conditions, applying 10 millisecond 4.0 volt pulses to their STM tip. Their imaging was performed at 0.8 volts, with a typical current of 0.3nA, though they imaged "in constant height mode", so either the current or voltage must have varied. Their STM tip was a mechanically cut Pt-Ir wire.
After applying their 4.0 volt pulses, the authors see substantially increased conductivity under their STM tip. They report I(V) curves before and after the pulse. Before the pulse, less than 1 nA flows for biases below 1 volt. After the pulse, the resistance near zero bias drops to ~500 megohms, and the current jumps steeply upwards at biases above ~0.25 volts, so the effective bandgap seems to have dropped. The authors attribute the gain in conductivity to polymerization of the cyano groups. The authors explicitly rule out the possibility that they had blown a hole through the film to the HOPG substrate. They did a control experiment where they used higher voltages and currents to expose the HOPG substrate, and they saw the normal atomic image of a graphite surface under those circumstances.
This authors demonstrated that they could write a 6 X 8 array of their marks in a 64 nm X 64 nm field, which was the objective of their current study. Presumably they will see how closely their marks can be placed without mutual interference in a later study.
The authors wrote that they were recording their marks "using single [crystal???] films". If these were single crystal films, it would be helpful to know the spacing of the cyano groups and their orientation relative to their STM’s electric field. This paper’s STM images do not display atomic resolution, and it would be useful to know if this is for fundamental reasons or only due to limitations in their current STM.
From the viewpoint of molecular nanotechnology, this technique could prove useful if it allows the fabrication of atomically precise wires in precise locations. The process must be quite anisotropic, given the formation of 0.8 nm marks in a 10 nm thick film, but it would be helpful to know more about the mechanism in order to see to what precision it can be pushed. Perhaps most crucially, we need to know if the polymerization is caused by the electric field or by the tunneling current. If it is field driven, then smaller electrodes (perhaps fullerene tubes or Tour wires) could impose more local fields. If it is current driven, then we are probably already at the physical limit, since their STM tip can image a graphite lattice.
Jeffrey Soreff is an IMM research associate.